









Indhold
Ehlers-Danlos syndrom
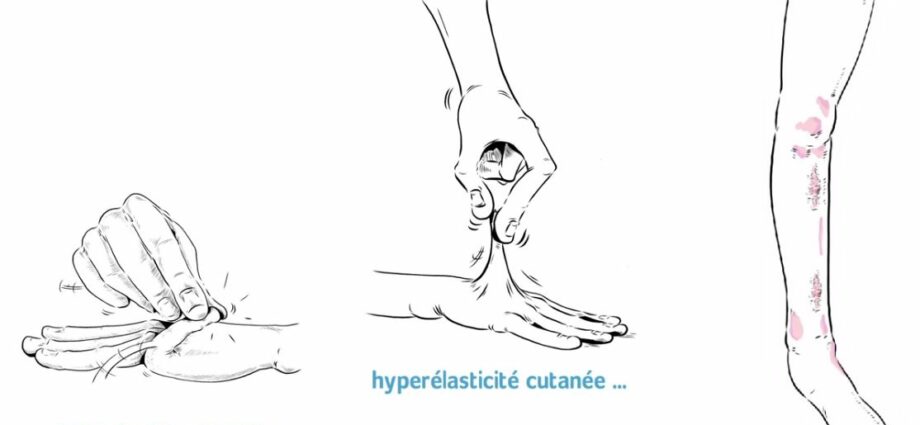
Le Ehlers-Danlos syndrom er en gruppe af genetiske sygdomme karakteriseret ved a bindevævs abnormitet, det vil sige understøttende væv.
Der er forskellige varianter af sygdommen1, de fleste har en hyperlaxitet i leddene, meget elastisk hud , skrøbelige blodkar. Syndromet påvirker ikke intellektuelle evner.
Ehlers-Danlos syndrom er opkaldt efter to medicinske hudlæger, den ene danske, Edvard Ehlers og den anden franskmand, Henri-Alexandre Danlos. De beskrev sygdommen igen i 1899 og 1908. |
Årsager
Ehlers-Danlos syndrom er en genetisk lidelse, der påvirker produktionen af kollagen, et protein, der giver elasticitet og styrke til bindevæv som hud, sener, ledbånd samt vægge i organer og organer. blodårer. Mutationer i forskellige gener (f.eks. ADAMTS2, COL1A1, COL1A2, COL3A1) ville være ansvarlige for de forskellige symptomer alt efter sygdommens forskellige former.
De fleste former for Ehlers-Danlos syndrom (EDS) arves af autosomalt dominerende tilstande. En forælder, der bærer mutationen, der er ansvarlig for sygdommen, har derfor 50% chance for at overføre sygdommen til hvert af deres børn. Nogle tilfælde opstår også ved spontane mutationer.
Komplikationer
De fleste mennesker med Ehlers-Danlos syndrom lever et relativt normalt liv, selvom de har begrænsninger på fysiske aktiviteter. Komplikationer afhænger af typen af involverede ADS.
- fordele ar vigtig.
- fordele kroniske ledsmerter.
- Tidlig gigt.
- Un aldring for tidlig på grund af sollys.
- Osteoporose.
Personer med EDS af vaskulær type (type IV SED) risikerer at få mere alvorlige komplikationer, såsom brud på vigtige blodkar eller organer såsom tarmen eller livmoderen. Disse komplikationer kan være dødelige.
Forekomst
Forekomsten af alle former for Ehlers-Danlos syndrom på verdensplan er cirka 1 ud af 5000 mennesker. type hypermobil, den mest almindelige, anslås til 1 ud af 10, mens vaskulær type, sjældnere, findes i 1 ud af 250 tilfælde. Sygdommen synes at påvirke både kvinder og mænd.