









Indhold
Bourneville tuberøs sklerose
Hvad er det ?
Bourneville tuberøs sklerose er en kompleks genetisk sygdom karakteriseret ved udviklingen af en godartet (ikke-kræft) tumor i forskellige dele af kroppen. Disse tumorer kan derefter lokaliseres i huden, hjernen, nyrerne og andre organer og væv. Denne patologi kan også forårsage alvorlige problemer i udviklingen af individet. Imidlertid varierer de kliniske manifestationer og sværhedsgraden af sygdommen fra patient til patient.
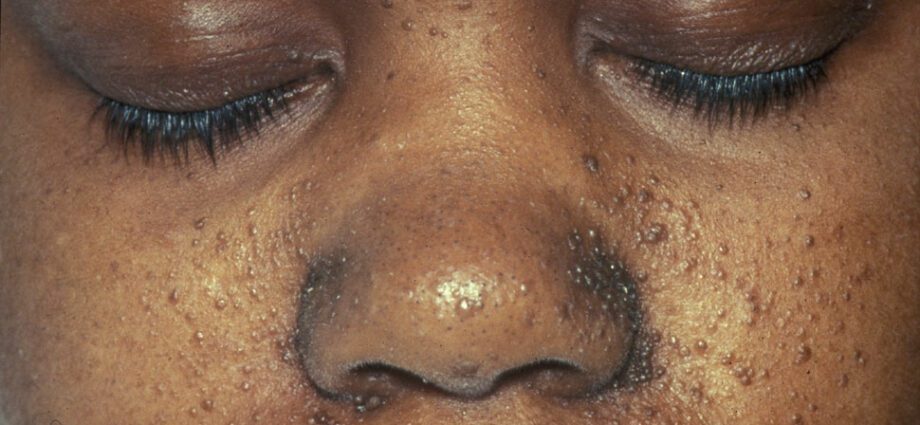
De tilknyttede hudabnormiteter ligner generelt pletter på huden eller områder, hvor huden er lysere end på resten af kroppen. Udviklingen af tumorer i ansigtet kaldes angiofibrom.
I forbindelse med hjerneskade er de kliniske tegn epileptiske anfald, adfærdsproblemer (hyperaktivitet, aggressivitet, intellektuelle handicap, indlæringsproblemer osv.). Nogle børn med sygdommen har endda en form for autisme, udviklingsforstyrrelser, der påvirker sociale interaktioner og kommunikation. Godartede hjernetumorer kan også forårsage komplikationer, der kan være dødelige for forsøgspersonen.
Udviklingen af tumorer i nyrerne er almindelig hos mennesker med tuberøs sklerose. Dette kan forårsage alvorlige komplikationer i nyrefunktionen. Derudover kan der udvikles tumorer i hjertet, lungerne og nethinden. (2)
Det er en sjælden sygdom, hvis udbredelse (antal tilfælde i en given befolkning på et givet tidspunkt) udgør 1/8 til 000/1 personer. (15)
Symptomer
De kliniske manifestationer forbundet med tuberøs sklerose i Bourneville varierer afhængigt af de berørte organer. Derudover varierer symptomerne forbundet med sygdommen meget fra individ til individ. Med symptomer, der spænder fra milde til svære.
De mest udbredte symptomer på denne sygdom omfatter epileptiske anfald, kognitive og adfærdsmæssige forstyrrelser, hudabnormiteter osv. De organer, der oftest rammes, er: hjernen, hjertet, nyrerne, lungerne og huden.
Udviklingen af ondartede (kræft) tumorer er mulig i denne sygdom, men er sjældne og påvirker hovedsageligt nyrerne.
De kliniske tegn på sygdommen i hjernen stammer fra angreb på forskellige niveauer:
– beskadigelse af corticale tuberkler;
– ependymale knuder (SEN);
– kæmpe ependymale astrocytomer.
De resulterer i: udvikling af mental retardering, indlæringsvanskeligheder, adfærdsforstyrrelser, aggressivitet, opmærksomhedsforstyrrelser, hyperaktivitet, obsessiv-kompulsive lidelser mv.
Nyreskade er karakteriseret ved udvikling af cyster eller angiomyolipomer. Disse kan føre til nyresmerter og endda nyresvigt. Hvis kraftig blødning er mærkbar, kan det være fra svær anæmi eller forhøjet blodtryk. Andre mere alvorlige, men sjældne konsekvenser kan også være synlige, især udviklingen af carcinomer (tumor i de celler, der består af epitelet).
Øjenskade kan ligne synlige pletter på nethinden, hvilket kan forårsage synsforstyrrelser eller endda blindhed.
Hudabnormiteter er talrige:
– hypomelaniske makuler: som resulterer i fremkomsten af lyse pletter på huden, hvor som helst på kroppen, som følge af en mangel på melanin, et protein, der giver farve til huden;
- udseendet af røde pletter i ansigtet;
– misfarvede pletter på panden;
– andre hudabnormiteter, afhængige fra individ til individ.
Lungelæsioner er til stede hos 1/3 af patienterne med en lille kvindelig overvægt. De tilhørende symptomer er så mere eller mindre alvorlige vejrtrækningsbesvær.
Sygdommens oprindelse
Sygdommens oprindelse er genetisk og arvelig.
Transmission involverer mutationer i TSC1- og TSC2-generne. Disse gener af interesse kommer i spil i dannelsen af proteiner: hamartin og tuberin. Disse to proteiner gør det muligt, gennem et interaktivt spil, at regulere celleproliferation.
Patienter med sygdommen er født med mindst én muteret kopi af disse gener i hver af deres celler. Disse mutationer begrænser derefter dannelsen af hamartin eller tubertin.
I den sammenhæng, hvor de to kopier af genet er muteret, forhindrer de fuldstændig produktionen af disse to proteiner. Denne proteinmangel tillader derfor ikke længere kroppen at regulere væksten af visse celler og fører i denne forstand til udviklingen af tumorceller i forskellige væv og/eller organer.
Risikofaktorer
Risikofaktorerne for at udvikle en sådan patologi er genetiske.
Faktisk er overførslen af sygdommen effektiv gennem en autosomal dominant tilstand. Enten er det muterede gen af interesse placeret ved et ikke-seksuelt kromosom. Derudover er tilstedeværelsen af kun én af de to kopier af det muterede gen tilstrækkelig til, at sygdommen kan udvikle sig.
I denne forstand har en person, der har en af disse to forældre, der lider af sygdommen, 50 % risiko for selv at udvikle den syge fænotype.
Forebyggelse og behandling
Diagnosen af sygdommen er først og fremmest differentiel. Det er baseret på atypiske fysiske kriterier. I de fleste tilfælde er de første karakteristiske tegn på sygdommen: Tilstedeværelsen af tilbagevendende epileptiske anfald og forsinkelser i forsøgspersonens udvikling. I andre tilfælde resulterer disse første tegn i hudpletter eller identifikation af en hjertetumor.
Efter denne første diagnose er yderligere undersøgelser afgørende for at validere diagnosen eller ej. Disse omfatter:
– en hjernescanning;
– en MR (magnetisk resonansbilleddannelse) af hjernen;
– en ultralyd af hjerte, lever og nyrer.
Diagnosen kan være effektiv ved barnets fødsel. Ellers er det vigtigt, at det udføres hurtigst muligt for hurtigst muligt at tage ansvar for patienten.
I øjeblikket er der ingen kur mod sygdommen. De tilknyttede behandlinger er derfor uafhængige af de symptomer, som hver enkelt person præsenterer.
Normalt gives antiepileptika for at begrænse anfald. Derudover er lægemidler til behandling af tumorceller i hjernen og nyrerne også ordineret. I forbindelse med adfærdsproblemer er specifik behandling af barnet nødvendig.
Behandling af sygdommen er normalt langsigtet. (1)