









Indhold
thalassæmi
Thalassæmi er et sæt af arvelige blodsygdomme, der påvirker produktionen af hæmoglobin (proteinet, der er ansvarlig for at transportere ilt). De varierer i sværhedsgrad: nogle forårsager ingen symptomer, mens andre er livstruende. En knoglemarvstransplantation overvejes i de mest alvorlige tilfælde.
Thalassæmi, hvad er det?
Definition af thalassæmi
Thalassæmi er karakteriseret ved en defekt i produktionen af hæmoglobin. Som en påmindelse er hæmoglobin et stort protein til stede i røde blodlegemer (røde blodlegemer), hvis rolle er at sikre transporten af dixoygen fra åndedrætssystemet til resten af kroppen.
Det siges, at thalassæmi er en sygdom i blodet. Røde blodlegemers transportfunktion er nedsat, hvilket kan have alvorlige konsekvenser for kroppen. På dette tidspunkt er det vigtigt at bemærke, at der er flere typer thalassæmi, der ikke har de samme karakteristika eller samme sværhedsgrad. Nogle har ingen symptomer, mens andre er livstruende.
Årsager til thalassæmi
Thalassæmi er genetiske sygdomme. De skyldes ændringen af et eller flere gener, der er involveret i syntesen af hæmoglobin, og mere præcist ændringen af de gener, der er involveret i produktionen af hæmoglobinproteinkæder. Der er fire af disse: to alfa-kæder og to beta-kæder.
Hver af disse kæder kan blive påvirket i thalassæmi. Vi kan også skelne:
- alfa-thalassæmi karakteriseret ved en ændring af alfa-kæden;
- beta-thalassæmi karakteriseret ved en ændring af beta-kæden.
Sværhedsgraden af alfa-thalassæmi og beta-thalassæmi afhænger af antallet af ændrede gener. Jo vigtigere det er, jo større er sværhedsgraden.
Diagnose af thalassæmi
Diagnosen thalassæmi stilles ved blodprøve. Den komplette blodtælling gør det muligt at vurdere udseendet og antallet af røde blodlegemer og dermed at kende den samlede mængde hæmoglobin. Biokemiske analyser af hæmoglobin gør det muligt at skelne alfa-thalassæmi fra beta-thalassæmi. Endelig gør genetiske analyser det muligt at evaluere antallet af ændrede gener og dermed at definere sværhedsgraden af thalassæmi.
Berørte personer
Thalassæmi er arvelige genetiske sygdomme, det vil sige, der overføres fra forældre til deres børn. De når hovedsageligt folk fra Middelhavsranden, Mellemøsten, Asien og Afrika syd for Sahara.
I Frankrig anslås forekomsten af alfa-thalassæmi til 1 ud af 350 mennesker. Forekomsten af beta-thalassæmi er anslået til 000 fødsler pr. 1 om året på verdensplan.
Symptomer på thalassæmi
Symptomerne på thalassæmi varierer meget fra sag til sag og afhænger hovedsageligt af graden af ændring af de gener, der er involveret i produktionen af hæmoglobinproteinkæder. Thalassæmi kan være symptomfri i deres mindre former og være livstruende i deres mere alvorlige former.
Symptomerne nævnt nedenfor vedrører kun de mellemliggende til større former for thalassæmi. Disse er blot hovedsymptomerne. Meget specifikke symptomer kan nogle gange ses afhængigt af typen af thalassæmi.
Anæmi
Det typiske tegn på thalassæmi er anæmi. Dette er mangel på hæmoglobin, som kan resultere i forekomsten af forskellige symptomer:
- træthed
- stakåndet;
- bleghed
- ubehag
- hjertebanken.
Intensiteten af disse symptomer varierer afhængigt af sværhedsgraden af thalassæmien.
Gulsot
Mennesker med thalassæmi kan have gulsot (gulsot), der er synlig på huden eller det hvide i øjnene.
Galdesten
Stendannelse inde i galdeblæren kan også ses. Beregninger er som "små småsten".
splenomegali
Splenomegali er en forstørrelse af milten. En af rollerne for dette organ er at filtrere blodet og filtrere skadelige stoffer, herunder unormale røde blodlegemer. Ved thalassæmi er milten stærkt mobiliseret og øges gradvist i størrelse. Smerter kan mærkes.
Andre, sjældnere symptomer
Mere sjældent kan alvorlige former for thalassæmi føre til andre abnormiteter. For eksempel kan det observeres:
- hepatomegali, det vil sige en stigning i leverens størrelse;
- knogledeformiteter;
- forsinket børns udvikling;
- sår.
Behandlingen af thalassæmi er afgørende for at begrænse forekomsten af disse komplikationer.
Behandlinger for thalassæmi
Behandlingen af thalassæmi afhænger af mange parametre, herunder typen af thalassæmi, dens sværhedsgrad og den pågældendes tilstand. De mest mindre former kræver ikke behandling, mens de alvorlige former kræver meget regelmæssig medicinsk overvågning.
Behandlingerne nævnt nedenfor vedrører kun de mellemliggende til større former for thalassæmi
Korrektion af anæmi
Når manglen på hæmoglobin er for stor, er regelmæssige blodtransfusioner nødvendige. De involverer at injicere den pågældende person med blod eller røde blodlegemer taget fra en donor for at opretholde et acceptabelt niveau af røde blodlegemer i blodet.
Vitamin B9 tilskud
Det kan anbefales at starte dagligt vitamin B9-tilskud, fordi behovet for dette vitamin er øget i tilfælde af thalassæmi. Vitamin B9 er involveret i produktionen af røde blodlegemer.
splenektomi
En splenektomi er kirurgisk fjernelse af milten. Denne operation kan overvejes, når anæmien er meget vigtig.
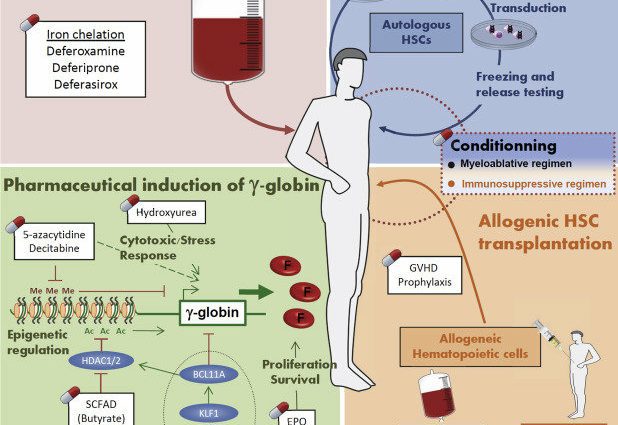
Behandling af jernoverskud
Mennesker med thalassæmi har en overbelastning af jern i deres krop. Denne ophobning kan føre til forskellige komplikationer. Dette er grunden til, at jernchelatorer tilbydes til at fjerne overskydende jern.
Knoglemarvstransplantation
En knoglemarvstransplantation er den eneste behandling, der permanent kan helbrede thalassæmi. Dette er en tung behandling, der kun tilbydes i de mest alvorlige former af sygdommen.
Forebyg thalassæmi
Thalassæmi er en arvelig genetisk sygdom. Der er ingen forebyggende foranstaltning.
På den anden side gør genetiske tests det muligt at påvise raske bærere (personer, der har et eller flere ændrede gener, men som ikke er syge). Et par raske bærere bør informeres om risikoen for at føde et barn med thalassæmi. I nogle tilfælde kan denne risiko vurderes af en genetiker. Prænatal diagnose kan også overvejes under visse betingelser. Det bør diskuteres med din læge.