









Indhold
Wilsons sygdom
Hvad er det ?
Wilsons sygdom er en arvelig genetisk sygdom, der forhindrer eliminering af kobber fra kroppen. Ophobning af kobber i leveren og hjernen forårsager lever- eller neurologiske problemer. Udbredelsen af Wilsons sygdom er meget lav, omkring 1 ud af 30 mennesker. (000) Der er en effektiv behandling af denne sygdom, men den tidlige diagnose er problematisk, fordi den forbliver tavs i lang tid.
Symptomer
Kobberophobning begynder ved fødslen, men de første symptomer på Wilsons sygdom vises ofte først i ungdomsårene eller i voksenalderen. De kan være meget forskellige, fordi flere organer påvirkes af akkumulering af kobber: hjerte, nyrer, øjne, blod ... De første tegn er hepatisk eller neurologisk i tre fjerdedele af tilfældene (henholdsvis 40% og 35%), men de kan også være psykiatrisk, nyre, hæmatologisk og endokrinologisk. Leveren og hjernen påvirkes især, fordi de allerede naturligt indeholder mest kobber. (2)
- Leverlidelser: gulsot, skrumpelever, leversvigt ...
- Neurologiske lidelser: depression, adfærdsforstyrrelser, indlæringsvanskeligheder, vanskeligheder med at udtrykke sig, rysten, kramper og kontrakturer (dystoni) ...
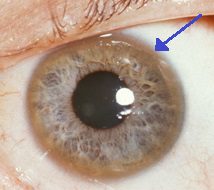
Keyser-Fleisher-ringen, der omgiver iris, er karakteristisk for ophobning af kobber i øjet. Ud over disse akutte symptomer kan Wilsons sygdom vise sig med ukarakteristiske symptomer, såsom generel træthed, mavesmerter, opkastning og vægttab, anæmi og ledsmerter.
Sygdommens oprindelse
Ved oprindelsen af Wilsons sygdom er der en mutation i ATP7B -genet placeret på kromosom 13, som er involveret i metabolismen af kobber. Det styrer produktionen af et ATPase 2 -protein, som spiller en rolle i transport af kobber fra leveren til andre dele af kroppen. Kobber er en nødvendig byggesten for mange cellefunktioner, men ud over kobber bliver det giftigt og skader væv og organer.
Risikofaktorer
Overførsel af Wilsons sygdom er autosomal recessiv. Det er derfor nødvendigt at modtage to kopier af det muterede gen (fra faderen og moderen) for at udvikle sygdommen. Det betyder, at mænd og kvinder er lige udsat, og at to forældre, der bærer det muterede gen, men ikke er syge, har en risiko for fire ved hver fødsel at overføre sygdommen.
Forebyggelse og behandling
Der er en effektiv behandling for at stoppe sygdommens progression og reducere eller endda fjerne dens symptomer. Det er også nødvendigt, at det startes tidligt, men det tager ofte mange måneder efter symptomernes begyndelse at diagnosticere denne tavse sygdom, lidt kendt, og hvis symptomer peger på mange andre tilstande (hepatitis, som er leverskade og depression for psykiatrisk involvering) .
En "chelaterende" behandling gør det muligt at tiltrække kobber og fjerne det i urinen og dermed begrænse dets ophobning i organerne. Det er baseret på D-penicillamin eller Trientine, medicin taget gennem munden. De er effektive, men kan forårsage alvorlige bivirkninger (nyreskade, allergiske reaktioner osv.). Når disse bivirkninger er for vigtige, ty vi til administration af zink, som vil begrænse optagelsen af kobber i tarmene.
En levertransplantation kan være nødvendig, når leveren er for beskadiget, hvilket er tilfældet for 5% af mennesker med Wilsons sygdom (1).
En genetisk screeningstest tilbydes søskende til en berørt person. Det giver anledning til en effektiv forebyggende behandling, hvis der påvises en genetisk abnormitet i ATP7B -genet.