









Indhold
Huntingtons sygdom
Hvad er det ?
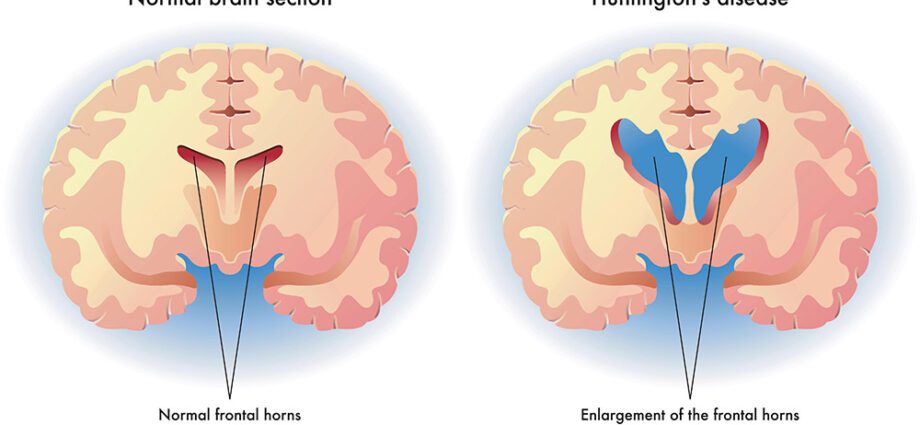
Huntingtons sygdom er en genetisk og arvelig neurodegenerativ sygdom. Ved at ødelægge neuroner i visse områder af hjernen forårsager det alvorlige motoriske og psykiatriske lidelser og kan føre til fuldstændigt tab af autonomi og død. Genet, hvis ændring forårsager sygdommen, blev identificeret i 90'erne, men Huntingtons sygdom forbliver uhelbredelig den dag i dag. Det rammer en ud af ti mennesker i Frankrig, hvilket repræsenterer omkring 10 patienter.
Symptomer
Det kaldes stadig nogle gange “Huntingtons chorea”, fordi det mest karakteristiske symptom på sygdommen er de ufrivillige bevægelser (kaldet choreic), det forårsager. Nogle patienter har imidlertid ikke choreiske lidelser, og symptomerne på sygdommen er bredere: til disse psykomotoriske lidelser tilføjes ofte psykiatriske og adfærdsforstyrrelser. Disse psykiatriske lidelser, der ofte forekommer ved sygdommens begyndelse (og sommetider vises før de motoriske lidelser) kan føre til demens og selvmord. Symptomer vises normalt omkring 40-50 år, men tidlige og sene former for sygdommen observeres. Bemærk, at alle bærere af det muterede gen en dag erklærer sygdommen.
Sygdommens oprindelse
Den amerikanske læge George Huntington beskrev Huntingtons sygdom i 1872, men det var først i 1993, at det ansvarlige gen blev identificeret. Det var lokaliseret på den korte arm af kromosom 4 og navngivet IT15. Sygdommen er forårsaget af en mutation i dette gen, der styrer produktionen af huntingtinproteinet. Den præcise funktion af dette protein er stadig ukendt, men vi ved, at den genetiske mutation gør det giftigt: det forårsager aflejringer i midten af hjernen, mere præcist i kernen af neuroner i caudatkernen, derefter i hjernebarken. Det skal dog bemærkes, at Huntingtons sygdom ikke systematisk er knyttet til IT15 og kan skyldes mutation af andre gener. (1)
Risikofaktorer
Huntingtons sygdom kan overføres fra generation til generation (det kaldes "autosomal dominant"), og risikoen for overførsel til afkommet er en ud af to.
Forebyggelse og behandling
Den genetiske screening af sygdommen hos personer med risiko (med en familiehistorie) er mulig, men meget under opsyn af lægen, fordi resultatet af testen ikke er uden psykologiske konsekvenser.
Prænatal diagnose er også mulig, men den er strengt indført ved lov, fordi den rejser spørgsmål om bioetik. Imidlertid har en mor, der overvejer en frivillig afslutning af graviditeten, hvis hendes foster bærer det ændrede gen, ret til at anmode om denne prænatal diagnose.
Til dato er der ingen helbredende behandling, og kun behandling af symptomer kan lindre den syge og bremse deres fysiske og psykiske forværring: psykofarmaka for at lindre de psykiatriske lidelser og de episoder med depression, som ofte går hånd i hånd med sygdommen. ; neuroleptiske lægemidler til at reducere choreiske bevægelser; genoptræning gennem fysioterapi og taleterapi.
Søgningen efter fremtidige terapier er rettet mod transplantation af føtale neuroner for at stabilisere hjernens motoriske funktioner. I 2008 beviste forskere fra Pasteur Institute og CNRS hjernens evne til at reparere sig selv ved at identificere en ny kilde til neuronproduktion. Denne opdagelse vækker nye håb om behandlingen af Huntingtons sygdom og andre neurodegenerative tilstande, såsom Parkinsons sygdom. (2)
Genterapiforsøg er også i gang i flere lande og bevæger sig i flere retninger, hvoraf den ene er at blokere ekspressionen af det muterede huntingtin -gen.